Getting started
You can learn how to use some of the HPG Pore most important features in this 5-minutes tutorial. It is assumed that you have HPG Pore installed, otherwise please visit the section Download and building.
To run this worked example you also need:
- A small FAST5 dataset (86MB): test_fast5.tar.gz
HPG Pore runs both:
- on a local environment, i.e., a single computer with a local or distributed POSIX file system, and
- on a Hadoop environment, a cluster of computers that implements the map-reduce paradigm.
The table below lists the available HPG Pore's commands and the environment (local or Hadoop) where it can run.
| HPG Pore command | Local | Hadoop |
|---|---|---|
| stats | yes | yes |
| events | yes | yes |
| signal | yes | yes |
| fasta | yes | yes |
| fastq | yes | yes |
| import | no | yes |
| export | no | yes |
| fast5names | no | yes |
To see all available commands and parameters, type:
$ ./hpp-pore.sh -h
Usage: hpg-pore.sh COMMAND
where COMMAND is one of:
stats explore Fast5 reads by computing statistics and plotting charts
signal plot the measured signal for a given Fast5 read
events extract events in text file for a given Fast5 read
fastq extract the sequences in Fastq format for a set of Fast5 reads
fasta extract the sequences in Fasta format for a set of Fast5 reads
fast5names extract all Fast5 names stored in the HDFS Hadoop file
Previous commands can run both on a local system and on a Hadoop environment (for the latter, use the option --hadoop.
Before executing those commands on a Hadoop environment, you must copy your Fast5 files to the Hadoop file system by running the command:
import copy the Fast5 files into the Hadoop environment (a HDFS Hadoop file)
To get back your Fast5 files imported to the Hadoop environment, use the command:e Hadoop file system by running the command:
export copy back your Fast5 files in the local filesystem from the Hadoop environment
Other commands:
version print the version
help print this help
Most commands print help when invoked w/o parameters.
Create a folder and download there the previous compressed FAST5 dataset, then uncompress this file:
$ mkdir ~/tutorial
$ cd ~/tutorial
$ tar zxvf test_fast5.tar.gz
Copy the HPG Pore script, the jar and dynamic library in the folder tutorial (where you have downloaded and uncompressed the dataset). So, you should have these files in that folder:
test_fast5
test_fast5.tar.gz
hpg-pore-0.1.0-jar-with-dependencies.jar
libhpgpore.so
hpg-pore.sh
Modify the LD_LIBRARY_PATH environment variable to include the path where the libhpgpore.so shared library is located, e.g.:
export LD_LIBRARY_PATH=~/tutorial:$LD_LIBRARY_PATH
To run the command stats, first you should create a folder where to save the output results:
$ mkdir out-stats
$ ./hpg-pore.sh stats --in test_fast5 --out out-stats
The command stats creates a file summary.txt containing several statistics and a folder per run to save histograms and graphs. In our example:
$ ls -ltr out-stats/
total 8
drwxrwxr-x 2 jtarraga jtarraga 4096 May 11 12:08 d5c085dc93da5740a906ccfd86aad93c2f0a44c8
-rw-rw-r-- 1 jtarraga jtarraga 1061 May 11 12:19 summary.txt
The content of the file summary.txt:
$ cat out-stats/summary.txt
-----------------------------------------------------------------------
Statistics for run d5c085dc93da5740a906ccfd86aad93c2f0a44c8
-----------------------------------------------------------------------
Template:
Num. seqs: 69
Num. nucleotides: 341458
Mean read length: 4948
Min. read length: 42
Max. read length: 17420
Nucleotides content:
A: 80450 (23.56 %)
T: 86374 (25.30 %)
G: 90780 (26.59 %)
C: 83854 (24.56 %)
N: 0 (0.00 %)
GC: 51.14 %
Mean read quality: 37
Complement:
Num. seqs: 26
Num. nucleotides: 144914
Mean read length: 5573
Min. read length: 830
Max. read length: 9544
Nucleotides content:
A: 35648 (24.60 %)
T: 36154 (24.95 %)
G: 37993 (26.22 %)
C: 35119 (24.23 %)
N: 0 (0.00 %)
GC: 50.45 %
Mean read quality: 37
2D:
Num. seqs: 20
Num. nucleotides: 136257
Mean read length: 6812
Min. read length: 1916
Max. read length: 10090
Nucleotides content:
A: 34325 (25.19 %)
T: 34088 (25.02 %)
G: 34143 (25.06 %)
C: 33701 (24.73 %)
N: 0 (0.00 %)
GC: 49.79 %
Mean read quality: 42
And the histograms and images generated by the the run d5c085dc93da5740a906ccfd86aad93c2f0a44c8:
$ ls -ltr out-stats/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 2216
-rw-rw-r-- 1 jtarraga jtarraga 122091 May 11 13:16 reads_per_channel.jpg
-rw-rw-r-- 1 jtarraga jtarraga 114092 May 11 13:16 yield_per_channel.jpg
-rw-rw-r-- 1 jtarraga jtarraga 143430 May 11 13:16 Template_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 136725 May 11 13:16 Complement_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 130031 May 11 13:16 2D_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 72637 May 11 13:16 Template_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 79887 May 11 13:16 Complement_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 72701 May 11 13:16 2D_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 99084 May 11 13:16 Template_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 100973 May 11 13:16 Complement_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 95601 May 11 13:16 2D_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 101578 May 11 13:16 Template_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 110502 May 11 13:16 Complement_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 103784 May 11 13:16 2D_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 118299 May 11 13:16 Template_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 135003 May 11 13:16 Complement_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 133907 May 11 13:16 2D_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 90795 May 11 13:16 Template_GC_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 91373 May 11 13:16 Complement_GC_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 101286 May 11 13:16 2D_GC_histogram.jpg
You can extract the sequences in format FastQ and FASTA by executing the commands fastq and fasta respectively, e.g.: extracting sequences in FastQ format:
$ mkdir out-fastq
$ ./hpg-pore.sh fastq --in test_fast5 --out out-fastq
A folder is created per run, in our case, we have one run: d5c085dc93da5740a906ccfd86aad93c2f0a44c8
$ ls -ltr out-fastq/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 1236
-rw-rw-r-- 1 jtarraga jtarraga 684625 May 11 14:28 template.fq
-rw-rw-r-- 1 jtarraga jtarraga 290472 May 11 14:28 complement.fq
-rw-rw-r-- 1 jtarraga jtarraga 273008 May 11 14:28 2D.fq
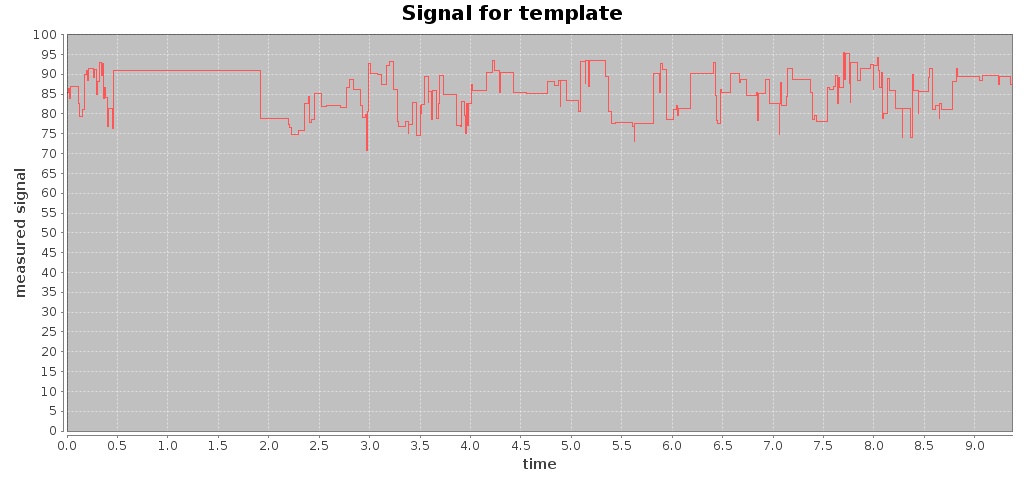
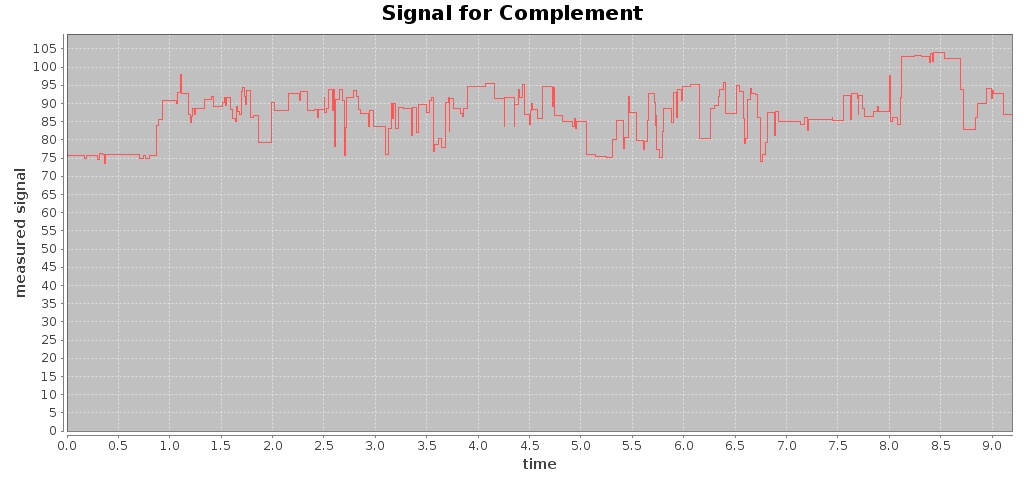
For a given Fast5 file you can also extract raw data of the electronic signal measured (by executing the command events) and plot the signal over time (by using the command signal), e.g. plotting the signal for the first 10 seconds:
$ mkdir out-signal
$ ./hpg-pore.sh signal --in test_fast5/LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5 --out out-signal --min 0 --max 10
For this Fast5 file, two signals are plotted for the template and complement sequences:
$ ls -ltr out-signal/
total 244
-rw-rw-r-- 1 jtarraga jtarraga 116307 May 11 14:14 template_signal.jpg
-rw-rw-r-- 1 jtarraga jtarraga 121762 May 11 14:14 complement_signal.jpg
About how to download and install Hadoop
Create a folder and download there the previous compressed FAST5 dataset, then uncompress this file:
$ mkdir tutorial
$ cd tutorial
$ tar zxvf test_fast5.tar.gz
Copy the HPG Pore script, the jar and dynamic library in the folder tutorial (where you have downloaded and uncompressed the dataset). So, you should have these files in that folder:
test_fast5
test_fast5.tar.gz
hpg-pore-0.1.0-jar-with-dependencies.jar
libhpgpore.so
hpg-pore.sh
Before processing your Fast5 files on a Hadoop environment, you have to import them to the Hadoop file system (HDFS), by invoking the command import:
./hpg-pore.sh import --in test_fast5 --out fast5_in_hdfs
You can check your files were copied to the HDFS by executing the Hadoop's command ls:
$ hadoop fs -ls fast5_in_hdfs
Found 2 items
-rw-r--r-- 3 hadoopuser1 hadoopuser1 89468918 2015-05-11 17:12 fast5_in_hdfs/data
-rw-r--r-- 3 hadoopuser1 hadoopuser1 203 2015-05-11 17:12 fast5_in_hdfs/index
The HPG Pore's command fast5names lists the filenames imported to the Hadoop system:
$ ./hpg-pore.sh fast5names --in fast5_in_hdfs
LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch13_file33_strand.fast5
LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch14_file33_strand.fast5
LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5
LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch163_file33_strand.fast5
...
...
These Fast5 names are useful to extract raw data of the electronic signal measured or to plot that signal from a given Fast5 file, e.g. plotting the signal for the first 10 seconds (on a Hadoop environment you must set the parameter --hadoop):
$ mkdir out-signal
$ ./hpg-pore.sh signal --in fast5_in_hdfs --fast5name LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5 --out out-signal --min 0 --max 10 --hadoop
As on the local environment, two images were generated:
$ ls -ltr out-signal/
total 236
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 115690 May 11 17:32 template_signal.jpg
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 120621 May 11 17:32 complement_signal.jpg
Extracting raw data of the electronic signal measured for the previous file:
$ mkdir out-events
$ ./hpg-pore.sh events --in fast5_in_hdfs --fast5name LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5 --out out-events/ --hadoop
$ ls -ltr out-events/
total 1968
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 419437 May 11 17:51 template_events.txt
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 583939 May 11 17:51 complement_events.txt
$ head out-events/template_events.txt
mean start stdv length model_state model_level move p_model_state mp_state p_mp_state p_A p_C p_G p_G p_Traw_index
85.384529 20407.621094 2.983408 0.018000 TTACT 145.747223 0 0.059361 TTACT 0.059361 0.361061 0.228866 0.190751 0.151697 0
86.403023 20407.638672 1.502944 0.009800 TACTC 146.695374 1 0.057656 TACTC 0.057656 0.289311 0.377167 0.170088 0.127892 1
83.816956 20407.648438 2.903588 0.011000 ACTCT 144.912308 1 0.138554 ACTCT 0.138554 0.216352 0.158154 0.152406 0.375754 2
86.954803 20407.660156 1.632599 0.072200 CTCTG 147.157684 1 0.204316 CTCTG 0.204316 0.228167 0.641092 0.090397 0.018639 3
82.601898 20407.730469 1.926103 0.012600 TCTGC 142.219894 1 0.174193 TCTGC 0.174193 0.018027 0.020496 0.114566 0.830867 4
The command export allows you to get back your Fast5 files from the Hadoop filesystem. Use the parameter fast5name to export a given Fast5 file, otherwise all your Fast5 files will be exported:
Exporting one single file:
$ mkdir out-export
$ ./hpg-pore.sh export --in fast5_in_hdfs --fast5name LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5 --out out-export/
$ ls -ltr out-export/
total 1816
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1856590 May 11 18:03 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5
Exporting all:
$ ./hpg-pore.sh export --in fast5_in_hdfs --out out-export/
$ ls -ltr out-export/
total 127976
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1318766 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch13_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1639952 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch14_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1856590 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 669484 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch163_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 2280863 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch166_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 857543 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch167_file33_strand.fast5
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1715957 May 11 18:05 LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch186_file33_strand.fast5
...
...
To run the command stats on a Hadoop system use the parameter --hadoop:
$ mkdir out-stats
$ ./hpg-pore.sh stats --in fast5_in_hdfs --out out-stats --hadoop
$ ls -ltr out-stats/
total 816
-rw-r--r--. 1 hadoopuser1 hadoopuser1 825198 May 12 11:59 raw.txt
drwxrwxr-x. 2 hadoopuser1 hadoopuser1 4096 May 12 11:59 d5c085dc93da5740a906ccfd86aad93c2f0a44c8
-rw-rw-r--. 1 hadoopuser1 hadoopuser1 1122 May 12 11:59 summary.txt
As commented, the file summary.txt contains statistics and the folder d5c085dc93da5740a906ccfd86aad93c2f0a44c8 contains the histograms and figures for that run.
And for extracting the sequences in format FastQ or FASTA on a Hadoop, use the commands fastq and fasta respectively with the parameter --hadoop:
$ mkdir out-fastq
$ ./hpg-pore.sh fastq --in fast5_in_hdfs --out out-fastq/ --hadoop
$ ls -tlr out-fastq/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 1224
-rw-r--r--. 1 hadoopuser1 hadoopuser1 273008 May 12 12:35 2D.fq
-rw-r--r--. 1 hadoopuser1 hadoopuser1 290472 May 12 12:35 complement.fq
-rw-r--r--. 1 hadoopuser1 hadoopuser1 684625 May 12 12:35 template.fq
$ mkdir out-fasta
$ ./hpg-pore.sh fasta --in fast5_in_hdfs --out out-fasta/ --hadoop
$ ls -tlr out-fasta/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 616
-rw-r--r--. 1 hadoopuser1 hadoopuser1 136711 May 12 12:36 2D.fa
-rw-r--r--. 1 hadoopuser1 hadoopuser1 145506 May 12 12:36 complement.fa
-rw-r--r--. 1 hadoopuser1 hadoopuser1 343029 May 12 12:36 template.fa