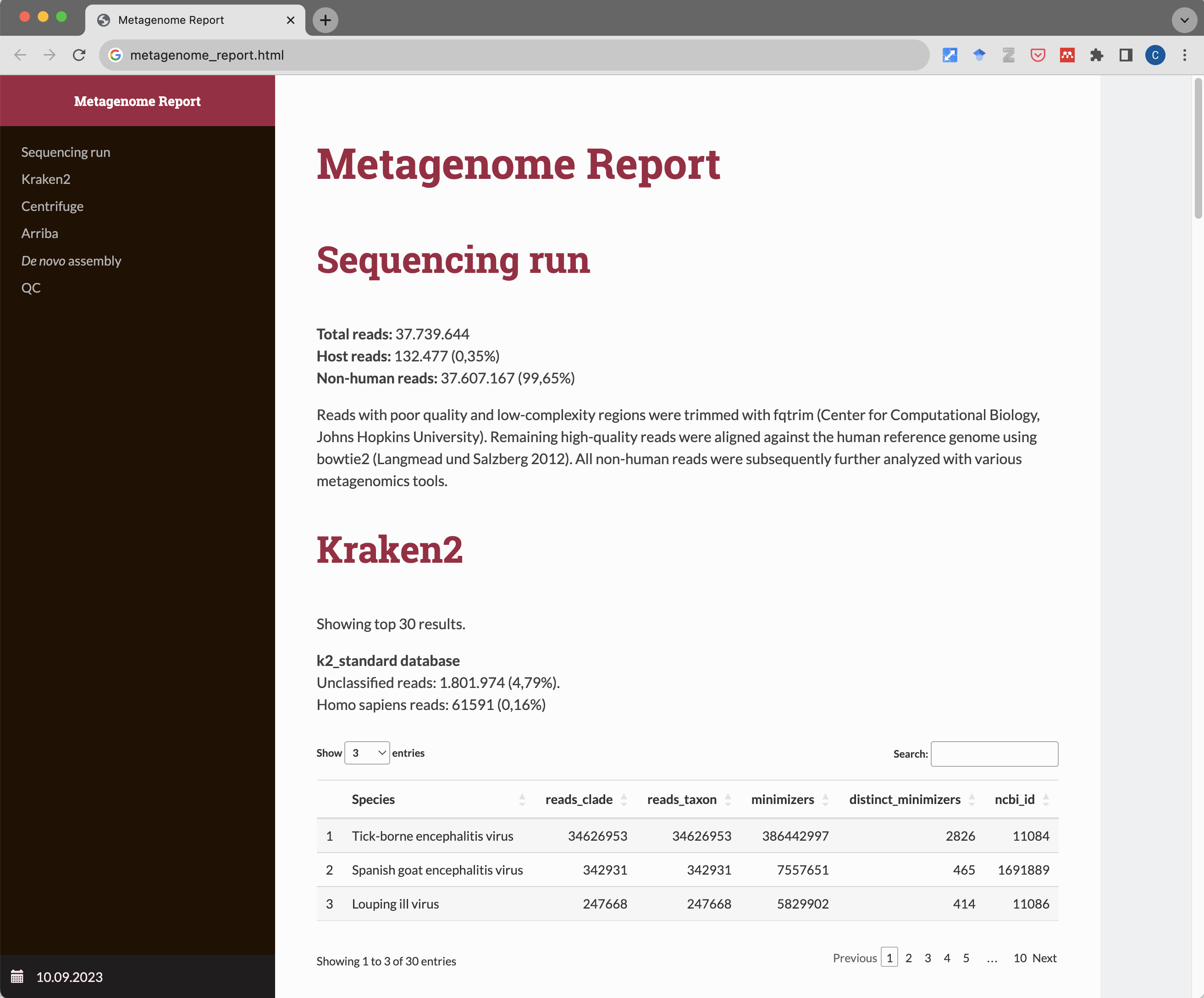
A pipeline for metagenomic sequencing experiments. Takes paired-end .fastq.gz files as input and generates a detailed HTML report with result tables. After quality trimming and filtering of low-complexity sequences with fqtrim, high-quality reads are being aligned to the human reference genome using Bowtie 2. Unaligned (non-human) reads are then subjected to several metagenomics tools.
First, taxonomic classification with Kraken 2 and Centrifuge using different reference databases (standard, viral, EUPATHDB48) is being performed. Next, reads are aligned against ~12,000 RefSeq virus genomes (as well as ~4,500 human-infecting virus strains related to the RefSeq viruses) and detection of viral integration sites into the human genome are detected using Arriba and STAR. In a last step, de novo assembly using MEGAHIT is being performed. Contigs > 1000 bp are automatically classified with Kraken 2 and Centrifuge. Output files can be used for manual downstream analyses such as BLAST or phylogenetic studies. All results are summarized in a comprehensive HTML report.
The pipeline is adapted to the SLURM job scheduler for parallel processing of multiple samples. Requires 140 GB memory (Centrifuge index with all non-redunandt NCBI sequences) and adjustable number of CPUs.
The following tools need to be installed and available in your $PATH:
- FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
- Fqtrim (https://github.com/gpertea/fqtrim)
- Bowtie 2 (https://github.com/BenLangmead/bowtie2)
- Kraken 2 (https://github.com/DerrickWood/kraken2)
- Centrifuge (https://github.com/DaehwanKimLab/centrifuge)
- STAR (https://github.com/alexdobin/STAR)
- Arriba (https://github.com/suhrig/arriba)
- MEGAHIT (https://github.com/voutcn/megahit)
Additionally, the following reference data is required:
- Bowtie 2 index of human genome (https://benlangmead.github.io/aws-indexes/bowtie)
- Kraken 2 indexes (standard, viral, EUPATHDB48) (https://benlangmead.github.io/aws-indexes/k2)
- Centrifuge index (NCBI: nucleotide non-redundant sequences) (https://benlangmead.github.io/aws-indexes/centrifuge)
- STAR index including viral sequences (can be downloaded using the arriba helper script
./download_references.sh hs37d5viral+GENCODE19, as described here https://arriba.readthedocs.io/en/latest/quickstart/)