| title | tags | authors | affiliations | date | bibliography | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cocktail Shaker: An open source drug expansion and enumeration library for peptides |
|
|
|
23 November 2019 |
paper.bib |
Without expensive software, the rapid creation and design of peptide ligand libraries has been a
challenge for many drug discovery scientists. Currently, protein- and peptide-based therapeutics constitute 10% of the
pharmaceutical market and will make up a larger proportion of the market in the future [@Craig:2013-1; @Bruno:2013-2]. With the demand for designing new peptide therapeutics on the rise, new high throughput peptide-specific informatic tools are needed. Currently, two platforms for this purpose exist: Molecular Operating
Environment (MOE) [@Reynolds:2010-3] and Rapid Peptides Generator (RPG) [@Maillet:2020-4] but both have drawbacks. MOE works efficiently for creating peptide molecule 3D chemical
files in one particular format (mol2), but at the high cost for a licence. RPG, although free of charge, does not account
for non-natural amino acids and production of multiple chemical files. In this study, I present the first open source python package,
Cocktail Shaker, developed for exploring, expanding, and synthesizing chemical peptide data.
Cocktail Shaker operates within the RDKit platform [@Landrum:2019-5] and is designed for the chemically-oriented
computational research community. RDKit utilizes C++-based functions for speed and rapid creation of molecule objects.
The toolkit offers a variety of utilities that includes: parsing and producing ready-to-use scientific files
designed for any chemical software, employing click chemistry methods for ease of exchange compounds, chemical data writing,
and chemical representation enumeration employed for machine learning.
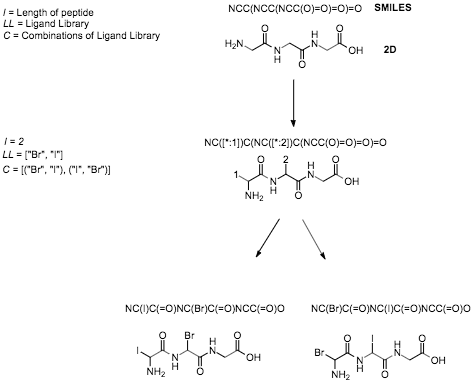
Cocktail Shaker consists of three major class objects available to the user: PeptideMolecule, CocktailShaker, and FileWriter.
Using string manipulation PeptideMolecule can build SMILES strings with allocated slots defined by the user. The user can
then enter the produced SMILES into the CocktailShaker object with a library of ligands represented by smiles and optional arguments
of whether to include generation of stereoisomers and/or natural amino acids. Cocktail Shaker will generate all combinations of the library and allocate them to a slot within the peptide. This process of
string manipulation is presented in Figure 1.
Cocktail Shaker also allows for File Writing of the molecules into a wide array of chemical formats (found in the documentation).
Cocktail Shaker uses RDKit to convert from 1D to 2D and the CIR Resolver built from webchem to convert 1D SMILES to 3D. At the request
of the user, the data is saved into one large data file or separated using the keyword fragmentation. This additional API
allows the user the flexibility to write a variety of files to implement in their respective chemical software.
Using Cocktail Shaker, individual research groups and companies can quickly construct private compound collections and progressively improve public
libraries with increased variations of chemical compound data.
Cocktail Shaker with its first version release provides a basis for drug expansion and enumeration. For future
releases Cocktail Shaker will be expanding into specifying shapes of compounds and, recently partnered with MolPort,
vendor information on any compound generated. It was presented at the RDKit UGM conference at the University of Hamburg
to the cheminformatics community with positive feedback with its second version 1.0.1. With incorporated feedback it will now be
released with version 1.1.8. With more contributions Cocktail Shaker will be an exciting tool for drug library creation
and drug discovery for scientists and engineers alike.
We acknowledge contributions from Ryland Forsythe as an academic consultation, Marvin Corro for quality assurance, Rose Gierth for technical documentation, and Elena Chow for her work on the graphics.